Vysokoteplotní palivové články, vhodná paliva a možnosti jejich využití (II)
4. Paliva pro vysokoteplotní palivové články
Vysokoteplotní palivové články jsou schopné používat jako palivo nejen vodík, ale také oxid uhelnatý, směsi CO s vodíkem, uhlovodíky, alkoholy apod. Vysokoteplotní články jsou zvláště citlivé na obsah prachu, par alkalických a těžkých kovů a na obsah síry/sloučenin síry v plynném palivu. Citlivost vůči amoniaku bývá menší. Uhlovodíky mohou být pomocí endotermní reakce s vodní parou za vyšších teplot (tzv. reformováním) převedeny na směs CO a vodíku. V systémech, které obsahují CO a vodní páru za vyšších teplot, běží také další reakce CO s vodou (viz rovnice 7). Také tuhá paliva (uhlí, biomasa) mohou být po zplynění 28 pomocí např. vodní páry za vyšších teplot přeměněna obecně na směs CO, H2 a uhlovodíků, které mohou být po odstranění dehtů, síry, amoniaku, těžkých kovů, apod. využity ve vysokoteplotních palivových článcích. Srovnání některých vlastností plynných a kapalných paliv využitelných ve vysokoteplotních palivových článcích je v Tabulce II.
| Palivo | Vodík | Metan | Metanol | Etanol | N-oktan |
| Molekulární hmotnost | 2,016 | 16,04 | 32,04 | 46,07 | 114,2 |
| Bod varu (°C) | -252,7 | -161,5 | 64,7 | 78,5 | 125,7 |
| Výparné teplo (kJ/kg) | 445,6 | 510 | 1100 | 855 | 368,1 |
| Spalné teplo při 25 °C (kJ/mol) | 241,8 | 802,5 | 638,5 | 1275,9 | 5512,0 |
| Hustota kapaliny (kg/m3) | 77 | 425 | 792 | 789 | 702 |
| Specifické teplo při 0 °C (J/mol K) | 28,8 | 34,1 | 76,6 | 112,4 | 188,9 |
| Teplota samovznícení (°C) | 571 | 632 | 470 | 362 | 220 |
| Meze výbušnosti ve vzduchu (% obj.) | 4 - 77 | 4 - 16 | 6 - 50 | 3 - 19 | 1 - 6 |
Tabulka II: Srovnání důležitých vlastností vybraných plynných a kapalných paliv1.
4.1 Plynná a kapalná paliva
Vodík je nejlepším palivem do vysokoteplotních palivových článků, protože je elektrochemicky reaktivnější než oxid uhelnatý nebo uhlovodíky. Jestliže jsou přiváděny do palivového článku ve směsi vodík a CO, je rychleji vyčerpáván vodík, tzn. že plyn odcházející z anody má relativně vyšší poměr CO/H2 než plyn na vstupu.
Plyny obsahující aspoň 10 - 20 % vodíku či oxidu uhelnatého již mohou být potenciálními palivy ve vysokoteplotních palivových článcích. Zemní plyn může být sice využíván také přímo 22,29 ve vysokoteplotních palivových článcích, ale sám o sobě (bez katalýzy) je poměrně málo reaktivní a má tendenci se za vysokých teplot částečně rozkládat na uhlík a vodík.
Na druhé straně, za teplotních podmínek provozu vysokoteplotních palivových článků a vzhledem k obvyklému složení materiálů anod (Ni-Cr-Al v případě článků MCFC a cermet s Ni v případě článků SOFC), je prakticky vždy aspoň část metanu přeměněna reakcí s anodovými produkty 30 (CO2 + H2O) také na vodík a CO. Provoz vysokoteplotních článků na "čistý" zemní plyn by byl teoreticky výhodný (viz Tabulka I), bohužel praktické zkušenosti ukázaly na snadný vznik uhlíku (sazí), na ničení cermetových anod vlivem tepelných napětí 1,30 (způsobených průběhem endotermních a exotermních reakcí) a na deaktivaci anod. Zvláště zbytky vyšších uhlovodíků v zemním plynu (etan, propan, butan apod.) jsou náchylné již za nižších teplot ke vzniku uhlíku. Niklové materiály elektrod mají tendenci ke tvorbě uhlíkatých úsad a deaktivaci 30,31.
| Palivo a produkt oxidace | T = 300 K | T = 1000 K | ||||
| ΔHf | ΔGf | η | ΔHf | ΔGf | η | |
| H2 → H2O(g) | -241,844 | -228,538 | 0,945 | -247,858 | -192,713 | 0,778 |
| CO → CO2 | -282,976 | -257,025 | 0,908 | -282,605 | -195,549 | 0,691 |
| CH4(g) → H2O(g) + CO2 | -802,264 | -800,839 | 0,998 | -800,466 | -800,812 | 1,000 |
| Butan(g) → H2O(g) + CO2 | -2656,948 | -2703,863 | 1,018 | -2661,814 | -2817,277 | 1,058 |
| Metanol(g) → H2O(g) + CO2 | -675,958 | -689,238 | 1,020 | -673,090 | -725,058 | 1,077 |
| Etanol(g) → H2O(g) + CO2 | -1277,640 | -1306,568 | 1,023 | -1277,902 | -1378,317 | 1,079 |
| C(s) → CO | -110,53 | -137,345 | 1,243 | -112,021 | -200,261 | 1,788 |
| C(s) → CO2 | -393,506 | -394,370 | 1,002 | -394,626 | -395,810 | 1,003 |
Tabulka I: Teoretické max. dosažitelné účinnosti η přeměn energie různých paliv vyjádřené jako spalovací nebo
reakční entalpie ΔHf (kJ/mol) na užitečnou práci vyjádřenou jako ΔGf (kJ/mol) za teplot 300 a 1000 K
(Thermochem. data - Barin I.7).
Proto je obvykle nutností zemní plyn (a hlavně jeho vyšší uhlovodíky) aspoň částečně přeměňovat (reformovat) reakcí s vodní parou nebo CO2 na reaktivnější a bezproblémovější plynná paliva - vodík a CO:
CH4 + H2O → CO + 3H2 (8)
CH4 + CO2 → 2CO + 2H2 (9)
Podobně jsou vodní parou reformovány i vyšší uhlovodíky - např. butan:
C4H10 + 4H2O → 4CO + 9H2 ( 10 )
Také nižší alkoholy (metanol, etanol) mohou být používány buď přímo po vypaření jako palivo nebo až po reformování vodní parou:
CH3OH + H2O → CO2 + 3H2 (11)
C2H5OH + H2O → 2CO + 4H2 (12)
Reakční tepla (entalpie) pro uvedené reakce probíhající při 300 a 1000 K jsou uvedená v Tabulce III. Po stránce spotřeby tepla je zvláště reformování metanolu energeticky nenáročné, probíhající za relativně nižších teplot a poskytující navíc ve vysokém výtěžku vodík. Proto se o metanolu uvažuje jako o možném palivu nejen pro vysokoteplotní, ale hlavně pro nízkoteplotní palivové články, které vyžadují čisté vodíkové palivo.
| Reformování (číslo rovnice) |
ΔHf (kJ/mol) | Spalné teplo produktů reformace při 1000 K (kJ) |
|
| 300 K | 1000 K | ||
| CH4 + H2O (8) | 206,244 | 225,713 | - 1026,179 |
| CH4 + CO2 (9) | 252,376 | 260,46 | - 1060,926 |
| C4H10 + 4H2O (10) | 651,552 | 699,328 | - 3361,142 |
| CH3OH + H2O (11) | 49,574 | 70,484 | - 743,574 |
| C2H5OH + H2O (12) | 255,688 | 278,74 | -1556,642 |
Tabulka III: Reakční tepla7 reformačních reakcí a spalná tepla7 produktů reakce po reformaci
vztažená na mol příslušného primárního paliva.
Srovnání Tabulky III s Tabulkou I ukazuje rozdíly ve spalných teplech (entalpiích) plynných paliv před reformováním a po něm. Všechny reformační reakce s vodou jsou endotermní, přičemž se vzrůstající teplotou roste jejich požadavek na dodání tepla. Při reformování je většinou přebytek vodních par, takže část CO reaguje dále na CO2.
4.2 Tuhá paliva
Přímá elektrochemická oxidace uhlíku v palivových článcích na CO nebo CO2 by byla energeticky velmi zajímavá, nabízející termodynamickou přednost ve změně Gibbsovy volné energie ΔGf dokonce větší než je entalpie reakce (viz Tabulka I). Tímto způsobem, protože změna entropie u oxidace pevného uhlíku je kladná, by se vlastně teplo dodané z vnějšku mohlo měnit na užitečnou práci 5. Ve skutečnosti je praktická realizace přímé elektrochemické oxidace uhlíku velmi obtížná. Při užití palivového článku typu MCFC by jako anoda musel sloužit přímo uhlík, který by byl ve styku s elektrolytem a kde by na rozhraní uhlíku a elektrolytu probíhala anodové reakce oxidace 4 na CO nebo CO2:
C + (CO3)2- → CO + CO2 + 2 e- (13)
C + 2(CO3)2- → 3CO2 + 4 e- (14)
Na anodu by se teoreticky nemusel přivádět žádný plyn a pouze by se odváděla plynná směs CO a CO2. Ve skutečnosti by se anodový prostor s porézní uhlíkovou anodou musel promývat inertním plynem (např. N2). Uhlíková anoda by samozřejmě musela být z relativně čistého uhlíku (bez síry, těžkých kovů a kovů alkalických zemin). Ještě složitější a také pravděpodobně méně účinné by bylo zařízení v případě článků typu SOFC, kde by muselo docházet ke kontaktu kompaktní uhlíkové anody s pevných elektrolytem, přičemž anoda by se spotřebovávala elektrochemickou reakcí. Dnes se spíše než o přímém využití tuhých paliv elektrochemickou reakcí uvažuje a prakticky zkouší kombinace zplyňování uhlí 1,33,34 nebo biomasy 28,35 a využití vzniklých plynů po náležitém vyčištění ve vysokoteplotních palivových článcích. U zplyňování uhlí závisí čistota produktů hlavně na teplotě a způsobu zplyňování.
Ze zplyňovacích systémů pro uhlí jsou dnes využívány tři základní typy 1:
Zplyňování v sesuvné vrstvě, vlivem protiproudu toku částic uhlí a plynu, má nevýhodu ve velkém obsahu dehtů a fenolických látek v plynech. Zplyňování ve fluidní vrstvě za teplot 800 - 950 °C je po této stránce lepší, avšak nejčistší plyn, prakticky bez dehtů, fenolů apod. je získáván ze zplyňování malých částic uhlí v úletovém režimu při teplotách nad 1200 °C. Zplyňování se provádí kontaktováním uhlí se směsí kyslíku a vodních par (tím se dostane vysoce výhřevný plyn s nízkým obsahem dusíku) nebo pomocí směsí vzduch-vodní pára. Při tomto způsobu generovaný plyn obsahuje obvykle nad 60 % dusíku.
Typické složení palivových plynů ze zplyňování v různých typech zařízení je uvedeno v Tabulce IV. Vzniklý uhelný, palivový plyn se musí zbavit sirovodíku, zbytků dehtů a také vyšších uhlovodíků, které by se mohly za nižších teplot rozkládat na uhlík. Integrace zplyňování uhlí, čištění plynů a vysokoteplotních palivových článků 33,34,36 je v pokročilém stadiu poloprovozního a provozního výzkumu. Velký důraz je kladen na efektivní a účinné čištění uhelného plynu. Zplyňování biomasy 28 a hlavně dřeva 34 je také zamýšleno k využití pro vysokoteplotní palivové články. Biomasa je při zplyňování velmi reaktivní. Pro vysokou rychlost reakcí stačí teploty již okolo 700 - 800 °C. Důležitým problémem je vyhnutí se vzniku sazí, hlavně při nižších teplotách zplyňování: k tomu bývá zapotřebí vyšší hmotový nebo molární poměr H2O/biomasa. Při zplyňování pomocí CO2 musí být tento analogický poměr (CO2/biomasa) asi dvakrát vyšší pro dosažení stejného účinku 28.
| Sesuvné lože O2 + H2O |
Fluidní vrstva O2 + H2O |
Úlet částic O2 + H2O |
Úlet částic vzduch + H2O |
|
| Uhlí: | Illinois No. 6 | Texas | Illinois No. 6 | Illinois No. 6 |
| Složení plynu | ||||
| Ar | Stopy | 0,7 | 0,9 | stopy |
| CH4 | 3,3 | 4,6 | 0,1 | 1,0 |
| C2H4 + C2H6 | 0,3 | |||
| H2 | 21,0 | 28,3 | 30,3 | 9,0 |
| CO | 5,8 | 33,1 | 39,6 | 16,0 |
| CO2 | 11,8 | 15,5 | 10,8 | 6,0 |
| N2 | 0,2 | 0,6 | 0,7 | 62 |
| NH3 | 0,4 | 0,1 | 0,1 | |
| H2O | 61,8 | 16,8 | 16,5 | 5,0 |
| H2S | 0,5 | 0,2 | 1,0 | ˜ 0,2 |
Tabulka IV: Typické složení uhelného plynu (% obj.) po zplyňování uhlí různými technologiemi1
Vyšší teploty zplyňování vedou jak u uhlí tak u biomasy k vyšším výtěžkům CO a nižším výtěžkům CO2, v případě vodíku bývá nalézána teplota (např. okolo 800 °C) s maximálním výtěžkem vodíku 28. Výhodou plynů ze zplyňování biomasy typu dřeva je velmi nízký až zanedbatelný obsah sloučenin síry a těžkých kovů. Obsah dehtů a oxidických a heterocyklických sloučenin však může být relativně vyšší.
Jako palivo pro VPČ se po vyčištění hodí i některé další bioplyny - např. z anaerobního čištění odpadních vod nebo skládkové plyny. Charakteristické pro tyto plyny je, že obecně obsahují jako hlavní složky metan a CO2, minoritní složky bývají různé, často sirovodík, čpavek. Bez náležitého čištění se ani tyto plyny nedají přímo použít ve vysokoteplotních palivových článcích. Vyšší obsah oxidu uhličitého (15 - 40 %) však není na překážku jejich využití.
4.3 Parní reformování paliv
Parním reformováním paliv (hlavně uhlovodíků a alkoholů - viz Tabulka III) se rozumí jejich převedení na jednodušší paliva (většinou hlavně směs CO, vodík, CO2) s vyšší výhřevností než mělo původní palivo. Parní reformování je běžně používaná metoda 37 výroby plynného paliva pro palivové články. Provádí se za teplot nad asi 500 °C, často za teplot 700 - 800 °C. Příklad složení plynů po reformování metanu přebytkem vodních par a změny ve složení plynů s teplotou jsou ukázány na obr. 7.
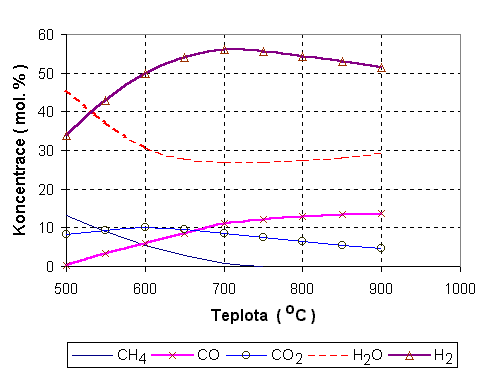
Obr. 7: Rovnovážné koncentrace plynů při reformování metanu vodní parou za teplot
nad 500 °C (P = 0,1 MPa). Uvažován přibližně dvojnásobný přebytek vodní páry1.
Reformování je silně endotermní reakcí a vyžaduje přívod tepla a vhodné katalyzátory. Jedním z nejdůležitějších problémů reformování je vyhnutí se produkci uhlíku (sazí) vlivem krakování a pyrolýzy uhlovodíků a rozkladu CO:
CH4 → C + 2H2 (15)
2CO → CO2 + C (16)
Endotermní pyrolýza metanu je významná za teplot asi nad 700 °C. Pyrolýza jiných uhlovodíků probíhá za nižších teplot. Exotermní rozklad CO je významný hlavně za nižších teplot, pod asi 600 °C. Tvorbě uhlíku se dá zabránit jednak přebytkem vodních par při reformování 37 a úpravou katalyzátoru, jednak tzv. před-reformováním vedoucím k odstranění vyšších uhlovodíků s velkou tendencí tvorby uhlíkatých úsad a snižujícím spotřebu tepla při vlastním vnitřním reformování 37. Niklové katalyzátory mají většinou tendenci podporovat vznik a usazování uhlíku. Proto je nutné je dopovat (např. alkalickými kovy apod.), nebo při silné tendenci k tvorbě sazí použít jiné katalyzátory (např. Ru na ZrO2 a Al2O3).
V případě VPČ není nutné odstraňovat CO, protože tento plyn je také využitelný. Vysokoteplotní palivové články dovolují dokonce provádění parního reformování uvnitř článků (internal reforming). Vnitřní reformování má dvě základní koncepce: přímé (DIR) a nepřímé (IIR) vnitřní reformování. V případě DIR se reformování děje přímo na anodě nebo v blízkosti anody. Teplo a pára potřebné pro reakci jsou dodávány přímo elektrochemickou reakcí. V případě IIR reformovací reakce probíhají v oddělené komůrce, která je ovšem v dobrém, hlavně tepelném kontaktu s palivovým článkem. Pára však není dodávána z palivového článku a také vznikající vodík není bezprostředně ve styku s anodou. Aplikování vnitřního reformování nabízí následující přednosti oproti vnějšímu reformování 25:
- Systémové náklady jsou menší, ušetří se přídavné zařízení
- U metody DIR je zapotřebí méně externí páry nebo dokonce žádná externí pára
- Rozdělení vodíku v palivovém článku u DIR je rovnoměrnější (je využita plocha anody)
- Bývá relativně vysoká konverze, protože vodík jako produkt reakce je odstraňován přednostně elektrochemickou reakcí.
Mezi nevýhody vnitřního reformování patří:
- Úprava anody na katalyzátor nebo další přídavný katalyzátor
- Katalyzátor se může deaktivovat nebo může být otráven (např. sírou, alkalickými kovy u MCFC apod.), může se sintrovat, může být vystaven teplotním stresům atd.
- Ačkoliv reformování je endotermní a elektrochemický proces je exotermní, jejich integrace může omezovat flexibilitu provozu palivového článku.
Vnitřní reformování paliv u MCFC
Anoda u těchto palivových článků je tvořena porézním niklem nebo speciálními slitinami na bázi niklu. Materiál anody musí mít dostatečné katalytické vlastnosti pro reformování, musí pracovat v korozivním prostředí s roztavenými alkaliemi z elektrolytu. Vyrovnanějšího teplotního pole se dosahuje recyklací anodového plynu 38, recyklací katodového i anodového plynu 38, předběžnou částečnou externí před-reformací uhlovodíkového paliva 1,30 a dokonce také řízenou mírnou deaktivací niklového katalyzátoru 39,40 (materiálu anody) plynným palivem obsahujícím do asi 60 ppm obj. síry. Hlavně se vždy jedná o úpravu katalyzátoru blízko vstupu plynného paliva do anodového prostoru nebo do prostoru reformační komůrky, protože pokles teploty, tepelné namáhání a deaktivace se projevují hlavně tam. Katalyzátor (anoda) může být proti účinkům alkalických tavenin, par a uhlíku chráněn povrchovou vrstvou platinového kovu (např. Ru, Rh), zlata, stříbra apod. Stárnutí niklového katalyzátoru se projevuje také sintrováním (hrubnutím zrn).
Vnitřní reformování paliv u SOFC
Vysokoteplotní články typu SOFC používají většinou nikl-keramických (Ni-ZrO2 cermetových) anod. Pro přímé reformování na anodě bývají dostatečně trvanlivé. Pro nižší teploty je také možné použít Ce-Ni cermetové katalyzátory dopované vápníkem. Vzhledem k typickým provozním teplotám nad 800 °C přímé vnitřní (DIR) reformování může probíhat na anodě, přídavného katalyzátoru není třeba. Aktivita katalyzátoru se může řízeně snižovat např. použitím menší koncentrace síry v plynu 39,40. Pro praktický provoz (hlavně u plynů obsahujících vyšší uhlovodíky) se doporučuje částečné externí reformování (okolo 40 - 60 % uhlovodíků), aby tepelné efekty při reformování zbytku paliva ve článku již nebyly tak velké a také aby nedocházelo k depozici uhlíku.
LITERATURA:
1. Larminie J., Dicks A.: Fuel Cell Systems Explained. Wiley, Chichester, 2001.
2. Appleby A.J., Foulkes F.R.: A Fuel Cell Handbook. 2.vyd., Kreiger , Huntington, 1993.
3. Haynes C.: J. Power Sources 92, 199 (2001).
4. Demin A., Tsiakaras P.: Int. J. Hydrogen Energy 26, 1103 (2001).
5. Peelen W.H.A., Hemmes K., de Wit J. H. W.: High Temp. Mater. Process. 2, 471 (1998).
6. Lutz A.E., Larson R.S., Keller J.O.: Int. J. Hydrogen Energy, 27, 1103 (2002).
7. Barin I. : Thermochemical Data of Pure Substances. 3. vyd. VCH, Weinheim, 1995.
8. de Haart L.G.J., Vinke I.C., Janke A., Ringel H. Tietz F.: v knize Solid Oxide Fuel Cells VII, H. Yokokawa, S.C. Singhal Ed., PV 2001-16, str. 111. The Electrochemical Society Proceedings Series, Pennington 2001.
9. Bosio B., Costamagna P., Parodi F., Passalacqua B.: J. Power Sources 74, 175 (1998).
10. Mathur A., Bali S., Balakrishnan M., Perumal R., Batra V.: Int. J. Energy Res. 23, 1177 (1999).
11. Lobachyov K.V., Richter H.J.: Energy Convers. Mgmt. 39, 1931 (1998).
12. Dicks A.L., Siddle A.: Assessment of Commercial Prospects of Molten Carbonate Fuel Cells. ETSU Report No. F/03/00168/REP, AEA Technology, Harwell, 1999.
13. Lobachyov K., Richter H.J.: J. Energy Resources Technol. 118, 285 (1996).
14. Lowrie F.L., Rawlings R.D.: J. Eur. Ceram. Soc. 20, 751 (2000).
15. McEvoy A.J.: J. Mater. Sci. 36, 1087 (2001).
16. Veyo S.E., Forbes C.A.: Proceedings of the 3 rd European Solid Oxide Fuel Cell Forum in Lausanne str. 79 (1998).
17. Biedenkopf P., Spiegel M., Grabke H.J.: Materials Corrosion 48, 488 (1997).
18. Kunz H.R.: J. Electrochem. Soc. 134, 105 (1987).
19. Yamamoto O : Elektrochim. Acta 45, 2423 (2000).
20. Tiffé E.I., Weber A,., Herbstritt D.: J. Eur. Ceram. Soc. 21, 1805 (2001).
21. Feng M., Goodenough J., Huank K., Milliken C.: J. Power Sources 63, 47 (1996).
22. Murray E.P., Tsai T., Barnett S.A.: Nature 400 , 649 (1999).
23. Das D., Edwards J., Kindermann L., Hilpert K., Putz G.: US patent 5 824 429, Int. Cl. H01M 4/90, 1998.
24. Isenberg H.O.: US patent 4 490 444, Int. Cl. H01M 8/12 , 1984.
25. Greiner H.: US patent 6 156 448, Int. Cl. H01M 8/12 , 2000.
26. Ringel H.: US patent 5 932 366, Int. Cl. H01M 8/12 , 1999.
27. Batawi E., Honegger K.: US patent 5 932 368, Int. Cl. H01M 4/86 , 1999.
28. Alderucci V., Antonucci P.L., Maggio G., Giordano N., Antonucci V.: Int. J. Hydrogen
Energy 19, 369 (1994).
29. Novikov G.I., Gamanovič N. M.: Ž. Prikl. Chim. 70, 1098 (1997).
30. Clarke S.H., Dicks A.L., Pointon K., Smith T.A., Swan A.: Catal. Today 38, 411 (1997).
31. Dicks A.L.: J. Power Sources 71, 111 (1998) .
32. Gür T.M., Huggins R.A.: US Patent 5 376 469, Int. Cl. H01M 8/12, 1994.
33. Wolk R.H., McDaniel J.: Energy Convers. and Manag. 33, 705 (1992).
34. Lobachyov K.V., Richter J.: Energy Convers. Mgmt. 38, 1693 (1997).
35. McIllveen D.R., Williams B.C., McMullan J.T.: Renewable Energy 19, 223 (2000).
36. Jansen D., Mozaffarian M.: Energy Convers. Mgmt. 38, 957 (1997).
37. Ahmed S., Krumpelt M.: Int. J. Hydrogen Energy 26, 291 (2001).
38. Kriechbaum K., Filip G.: US patent 6 136 462, Int. Cl. H01M 8/04, 2000.
39. Nielsen J.R., Christiansen L.J., Petersen K.A.: US patent 5 688 609, Int. Cl. H01M 8/10,
1997.
40. Hsu M.S.: US patent 5 747 185, Int. Cl. H01M 8/02, 1998.
41. Bevc F.: Proc. Inst. Mech. Engrs. 211, Part A , 359 (1997).
42. Hassmann K., Heidug W.K., Veyo S.: Brennstoff-Wärme-Kraft 51 , 40 (1999).
43. Wolfe D., Minh N., Meister K., Matulich D.: US patent 5 968 680, Int. Cl. H01M 8/04,
1999.
44. StockA.: US patent 6 124 050, Int. Cl. H01M 8/04, 2000.